Proof of Concept: DEL for PROTACs
DNA-Encoded Chemical Library Screening Is a Great Tool for Identifying Novel Binders Suitable for Bispecific Degraders
The Value of DEL for PROTACs and Other Bispecifics
Proteolysis targeting chimeras (PROTACs) are bispecific molecules that induce protein degradation of a specific target. These compounds have been a hot area of chemistry and pharma since they were discovered around 20 years ago.
Because of their structure, PROTACs have long been considered the perfect application for DNA-Encoded Library (DEL) screening. PROTACS have modular structures, consisting of a target binding moiety, a linker, and an engager with an E3 ligase. What makes DEL perfect for creating this bispecific structure is that in the library, the linkage point is built in already, connecting the discovered compound to its DNA label. In fact, you even know that the linker is tolerated in the binding event because it was there when you discovered the interaction. So all you need to do is replace the DNA with an appropriate E3 ligase engager and your discovery is ready to go.
Discovery of Bispecific Estrogen Receptor α Degraders With Novel Binders
To demonstrate this concept, our team built a bispecific degrader (PROTAC) of estrogen receptor α (ERα) with the goal of improving upon current modulators of ERα signaling (aromatase inhibitors/SERMs/SERDs) that are used to treat ER+ breast cancer. (For the full details, see our article, Bispecific Estrogen Receptor α Degraders Incorporating Novel Binders Identified Using DNA-Encoded Chemical Library Screening just out in the Journal of Medicinal Chemistry.)
We screened around 120 billion DNA-encoded molecules against wild type ERα andthreegain-of-function (GOF) mutants conferring partial resistance to existing endocrine therapies (Fig. 1a). Once enriched small molecules were identified via their DNA sequences, the small molecule binders were quickly leveraged in a parallel click chemistry array producing PROTACs for ERα with a variety of E3 binders (Fig. 1b).
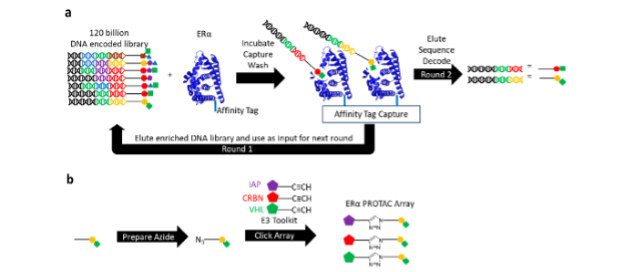
Reprinted with permission from Disch J.S. et al., Journal of Medicinal Chemistry 2021 64 (8), 5049-5066, DOI: 10.1021/acs.jmedchem.1c00127. Copyright © (2021) American Chemical Society.
Simultaneous Screening Yields Two Potent Off-DNA Compounds Enriched Against the Wild Type and All Three GOF Mutants
Because DEL library selection delivers not just hits, but profiles of hits for multiple targets included in the experiment, it saves significant time. In this case, the primary discovery screen identified two potent compounds that were enriched against the wild type ERα and three of the most common mutants — S463P, Y537S, and D538G (Fig. 2a & b). Additionally, the two compounds were not enriched in the presence of estradiol, indicating that they likely bind the estradiol binding pocket. To confirm the selection output, direct off-DNA analogs of the two potent compounds were synthesized, and their binding affinity was assessed in a fluorescence polarization binding assay (Fig. 2c).
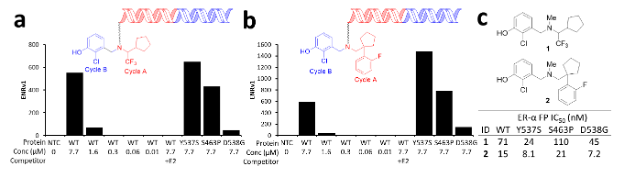
Reprinted with permission from Disch J.S. et al., Journal of Medicinal Chemistry 2021 64 (8), 5049-5066, DOI: 10.1021/acs.jmedchem.1c00127. Copyright © (2021) American Chemical Society.
Ensuring That the New Compounds Are Binding Antagonistically, Not Agonistically
Even though the goal of this bispecific therapy is to bind and then elicit degradation, it’s important to remember that the small molecule binder could have a cellular effect on its own. In this case the binders we discovered turned out to be agonists when dosed into cells. We were able to convert our binders into antagonists, making small changes to the structure to build a Structure Activity Relationship (SAR) that distinguished antagonism from agonism.
With click chemistry on our most active antagonists, we produced a series of bispecific compounds with variable E3 binding elements and variable linker lengths. Testing for ERα degradation in human breast cancer cells (MCF7) revealed two promising candidate PROTACs: 18 and 21. These two compounds engage VHL as the E3 component; compounds engaging IAP and CRBN did not demonstrate comparable degradation. Both 18 and 21 demonstrated potent binding of wild type ERα and all three GOF mutants (Fig. 3), degraded ERα, led to MCF7 viability defects and downregulated PGR expression.
Despite the low permeability, limited solubility, and high plasma protein binding of 18 and 21, these two PROTACs achieved significant plasma exposures when dosed subcutaneously (Fig. 3f), which warranted evaluation of their ERα degradation activities in vivo.
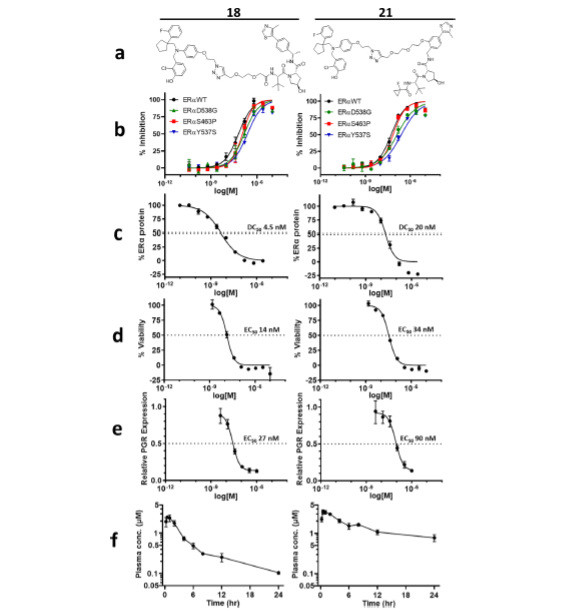
Reprinted with permission from Disch J.S. et al., Journal of Medicinal Chemistry 2021 64 (8), 5049-5066, DOI: 10.1021/acs.jmedchem.1c00127. Copyright © (2021) American Chemical Society.
Testing Tumor Knockdown In Vivo
We compared the efficacies of compounds 18 and 21 with that of standard-of-care Fulvestrant in a wild type, estrogen-dependent MCF7 xenograft model (Fig. 4). Compound 21 decreased tumor volume almost as well as Fulvestrant. These results demonstrate that 21 is excellent for inhibition and degradation of ERα, both in vitro and in vivo. The ERα-binder of 21 was identified directly from the DEL screen with minimal optimization.
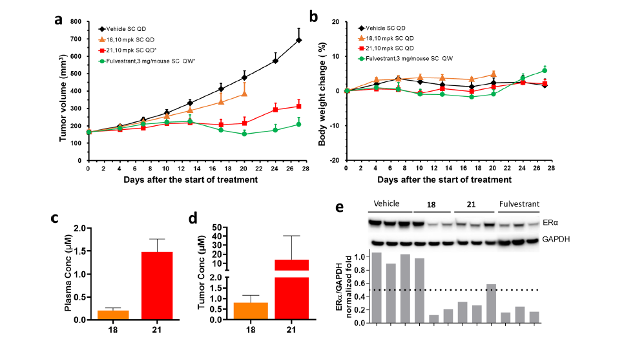
Reprinted with permission from Disch J.S. et.al., Journal of Medicinal Chemistry 2021 64 (8), 5049-5066, DOI: 10.1021/acs.jmedchem.1c00127. Copyright © (2021) American Chemical Society.
X-Chem Can Help You Identify and Assemble the Binders, Linkers, and E3 Ligands for Your Next PROTAC Project
This case demonstrates the potential and power of DEL for PROTAC applications. As the leader in DEL technology, X-Chem can help you screen DNA-encoded chemical libraries to identify novel binders as we did here and set you on the path to discovery of a groundbreaking compound. From hit identification for challenging targets to biochemical and biophysical profiling and elucidation of SAR through medicinal and synthetic chemistry, we’re here to assist you in your quest to unearth tomorrow’s life-saving therapies. Explore our website to learn more.
Reference
- Disch, J.S. et al. Bispecific Estrogen Receptor α Degraders Incorporating Novel Binders Identified Using DNA-Encoded Chemical Library Screening. Journal of Medicinal Chemistry 2021, 64 (8), 5049-5066.
2024: A Year of Scientific Excellence for X‑Chem
One of the best parts of being part of X-Chem is seeing all the great science being conducted around the...
The Importance of Selecting the Right CRO Partners for Drug Discovery Success
When it comes to drug discovery and development, having the right team and partners is critical for success. For a...